New paper out: (a bit delayed due to travelling the world for science…)
Nature Genetics 2015 Jun;47(6):632-9. doi: 10.1038/ng.3281
This paper provides a whole-genome snapshot of nearly 2000 genomes of the typhoid bacterium, Salmonella Typhi. The strains involved come from 63 countries contributed by dozens of people around the world, and were sequenced at the Sanger Institute with funding from the Wellcome Trust.
You can get the raw sequence reads under accession ERP001718, and play with the phylogenetic tree and associated map at the new MicroReact website:

Countries included in the study
I will post more later about plotting trees & metadata dynamically with MicroReact, and statically with Python and R.
But back to typhoid. This project is special for me for a number of reasons…
- It is about Typhi, the bug that suckered me into directing my genomics skills into studying pathogens and infectious disease, and was the subject of my PhD project with Gordon Dougan and Julian Parkhill at the Sanger Institute, and Duncan Maskell at Cambridge.
- It is a natural continuation of my PhD project, with the grunt work done by the new PhD student who took over typhoid genomics work in the Dougan lab when I moved back to Australia (Vanessa Wong, MD PhD), with me helping to direct the analysis from down here in Melbourne.
- It is a great illustration of how sequencing has changed… The first Typhi genome sequence was done at the Sanger Institute using capillary sequencing, and was published in 2001 (Parkhill et al, Nature). In my PhD project (also at the Sanger Institute), I analysed 19 Typhi genomes sequenced with two sequencing platforms that were new and super-duper back in 2006: 454 (now dead) and Solexa (now known as Illumina – currently ruling the sequencing world globally). This was published in 2008, seven years after the first genome (Holt et al, Nature Genetics). Now, another 7 years later in 2015, we are publishing almost 2000 genomes.
- Typhi is still one of the best examples I know of how sequencing has transformed bacterial surveillance and opened up a whole new field of genomic epidemiology. Before we could look at whole genomes, every Typhi strain looked pretty much the same genetically… there is so little variation, that lower resolution approaches like MLST just couldn’t tell us anything. Now that we can capture whole genomes relatively easily, we can track the transmission and evolution of these bugs essentially in real time.
So what did all this sequencing achieve? Basically we learnt a lot about a particularly tricky clone, called H58, that has spread quite rapidly across Asia and Africa and is responsible for most cases of multidrug resistant typhoid (infections that don’t respond to treatment with most antibiotics). About half of all our isolates belonged to this clone.
- By comparing root-to-tip branch lengths in the phylogenetic tree of H58 to the isolation dates of each strain, we found evidence of a temporal signal. So we did BEAST analysis, using the isolation date of each strain to date the tips and model mutation rates and divergence dates for H58. This showed that SNPs accumulated slowly in the Typhi H58 genome, at a rate of ~2 SNPs every 3 years. This placed the emergence of H58 at ~1989, just before our oldest example of H58 (1992). We haven’t been able to do proper dating in Typhi before, probably because most of the samples we’ve looked at previously have been phylogenetically diverse strains that are separated by centuries of evolution including periods of epidemic transmission (higher mutation rate per unit time) and long-term carriage (lower mutation rate per unit time). Here we probably have enough data from a period of epidemic transmission of H58 that the signal from epidemic transmission is detectable. I think this is very similar to Mycobacterium tuberculosis (TB), which notoriously has very little temporal signal, and yet a localised 4-decade transmission chain in Argentina showed very strong temporal signal.

- The geographical distribution of the H58 isolates tell us a lot about the routes by which H58 has travelled the world. The tree of H58 is so big that it’s hard to see what’s happening…. so to make it easier, I used R to collapse localised subclades of H58 that contained isolates from a single country (panel A – the size of the circle reflects the number of isolates in the subclade), and showed the time span for each subclade next to the tree (panel B). Occasionally there were one or two isolates within a localised subclade that were sourced from neighbouring countries, indicating transfer to those countries… these are shown in panel C.
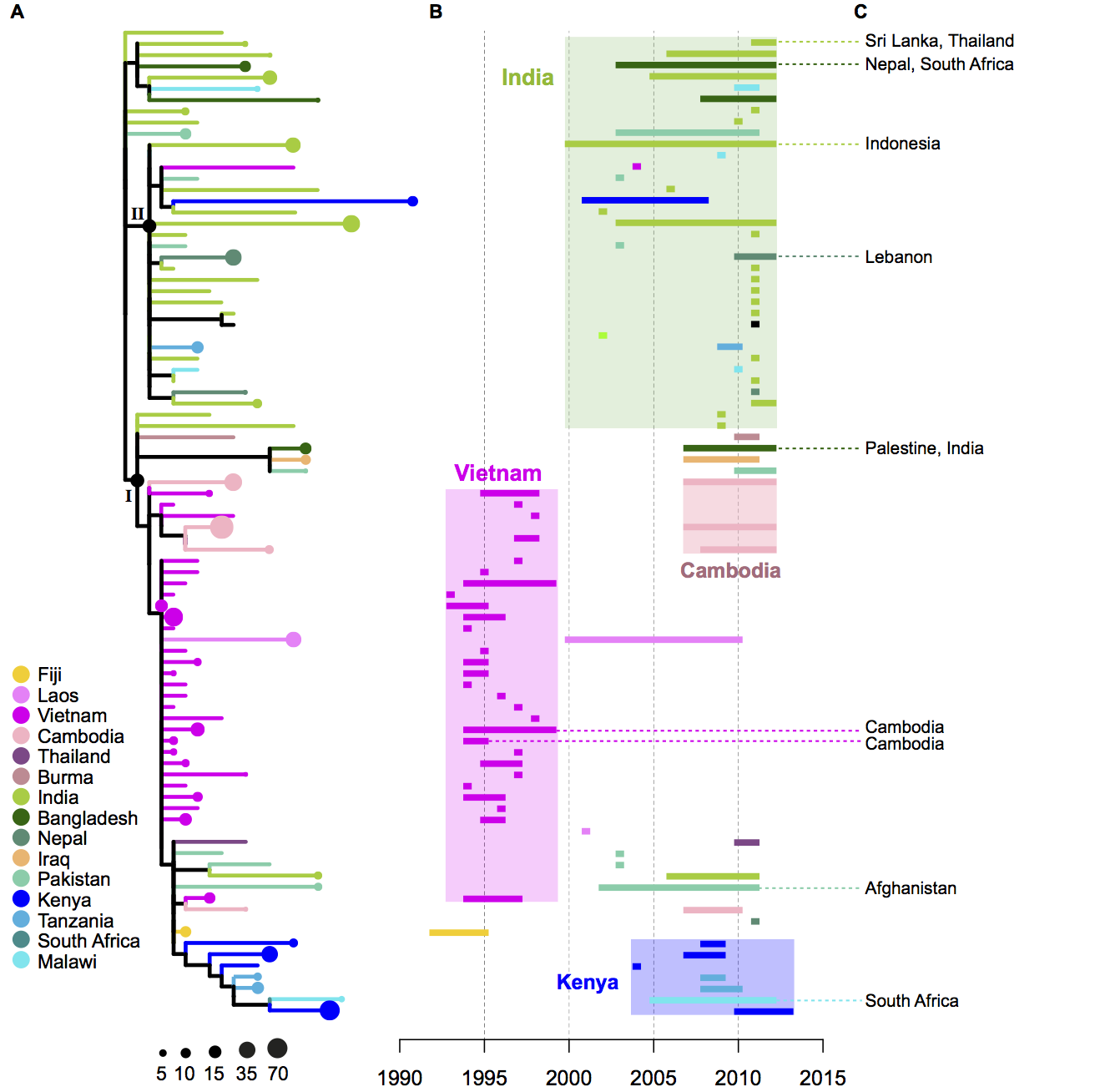
- We inferred these geographical patterns of the spread of H58, based on the tree and the regions of isolation:

- We learnt a lot about the evolution of multidrug resistance in Typhi H58. We knew that resistance to all first-line antibiotics was usually encoded in one big transposon, which came into H58 in a IncHI1 plasmid. But the new collection showed that this transposon has transferred into the Typhi H58 chromosome, not once but many times! These transfers have happened in separate events, in different parts of the world, and into different parts of the chromosome. This is what the transposon looks like, and two of the insertion sites relative to the reference chromosome (CT18):

- Finding transposon insertion sites is tricky! The transposon has copies of the IS1 transposase at either end, which we think are responsible for moving the whole transposon around. This poses a problem for genome assembly with short reads. One way around this is to sequence with long reads… the figure above shows two different insertion sites that we confirmed by using PacBio sequencing to get complete genomes. But we had >850 H58 genomes sequenced using Illumina which gives us short reads, so really we needed to figure out the insertion sites as best we could using the Illumina data. Luckily my PhD student Jane Hawkey had been working on a method to do this, called ISMapper. Using this approach, we could identify all the IS1 insertion sites in every Illumina-sequenced genome. We also found a couple of additional plasmids. This is where all the different multidrug resistance determinants are in the H58 population:

Finally, a clear message from this study is that we need to do a lot more sequencing of Typhi! While we have a lot of genomes here, there are large geographic areas that we just don’t know much about. Plus, we have seen that antibiotic resistance is evolving and changing fast, and we will need to keep up with this using ongoing genomic surveillance.