Today I’m pleased to see the final version of our paper on antimicrobial resistance in intestinal E. coli from Asian & African children published in Nature Microbiology. This is last piece of the puzzle from Danielle Ingle’s PhD research, a tremendous effort centred around the analysis of a collection of ~200 atypical enteropathogenic E. coli (aEPEC) isolated from cases and controls in seven countries during the Global Enterics Multicentre Study (GEMS).
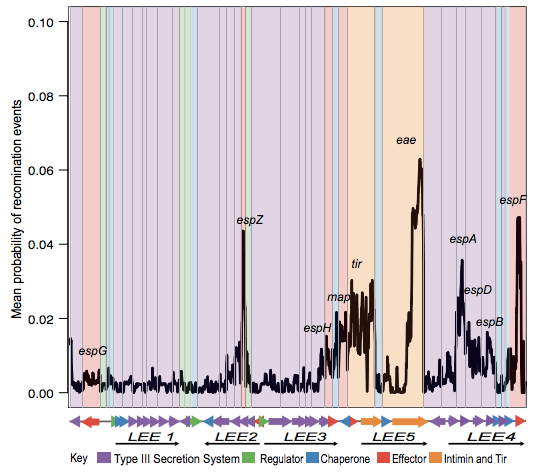
The first analysis of the genome data from this collection was reported in this 2016 paper, also in Nature Microbiology. It focused on understanding the population structure of the pathotype, including establishing a framework for looking at variation in the primary virulence locus (the LEE pathogenicity island; see blog post here).

Danielle then looked at serotype diversity in the collection, and used the experience to tackle the problem of O and H serotype prediction from genome data. That work is detailed in this Microbial Genomics paper, which utilises the phenotypes and genome data from the GEMS aEPEC collection to assess the reliability of predictions.
Finally we turned our attention to antimicrobial resistance (AMR) in the isolate collection – characterising resistance phenotypes, looking at known genetic determinants of AMR in the genome data, and also examining data on prescribing of antimicrobials for treatment of diarrhoeal disease in children at each study site.
So what did we find?
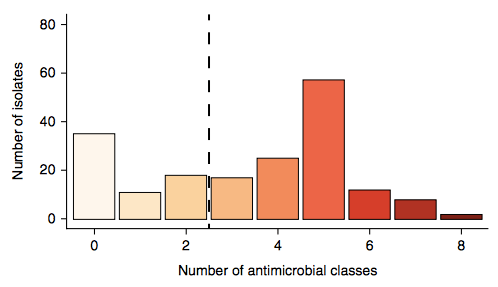
Firstly, whether we consider AMR phenotypes or genotypes we see that AMR was rampant, with most strains either multidrug resistant (65%; resistan to ≥ 3 drug classes) or susceptible to all drugs tested (19%):
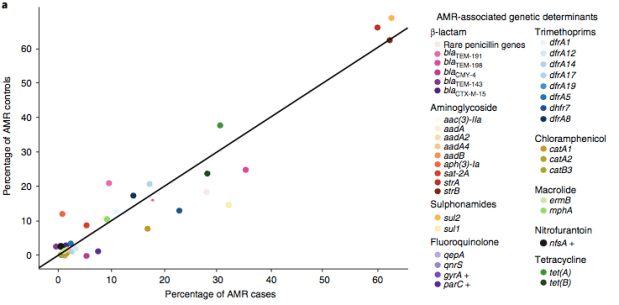
We found >40 different acquired AMR genes in the genomes, and also point mutations that are known to be associated with resistance to fluoroquinolones (in gyrA, parC) or nitrofurantoin (nfsA). Notably there was no difference between AMR rates in cases and controls, even at the level of individual genes:
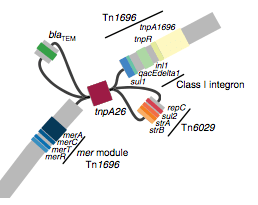
We found that many of these AMR genes co-occured together in known mobile genetic elements:

Quite often the structures of these elements were not totally resolvable from the genome assemblies, which were based on short Illumina reads only (no long reads for this data set unfortunately!)… but nevertheless, Danielle could often resolve co-localisation of these genes from the assembly graphs using Bandage:
We had seen in the first paper that the isolates were highly diverse, comprising dozens of distinct clones… this tree is inferred from a core gene alignment of the study isolates together with some other genomes for context (GEMS study isolates are indicated as dark blue in the outer ring). The ten shaded clades indicate dominant clonal groups in the study population.

Back to the AMR study. We did a discriminant analysis of principle components (DAPC) to see whether the variation in the distribution of genetic determinants amongst the genomes could be used to discriminate between the clonal groups, and saw that AMR was not associated with individual clones:
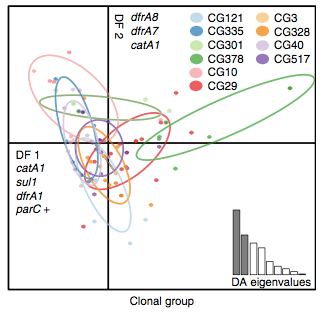
Instead we found that variation in AMR gene complement could discriminate isolates from different geographical region, suggesting that AMR genes more often reflect horizontal acquisition from distinct local gene pools in different parts of the world, rather than fixed features of their host bacterium that travel the world with their host strain (clone):

In particular, we saw that fluoroquinolone resistance associated mutations in gyrA were associated with Asian sites; while sites in East vs West Africa could be discriminated by the presence of different dihydrofolate reductase (dfr) genes responsible for trimethoprim resistance, with dfrA8 being more common in West Africa and dfrA5 being present in East Africa.
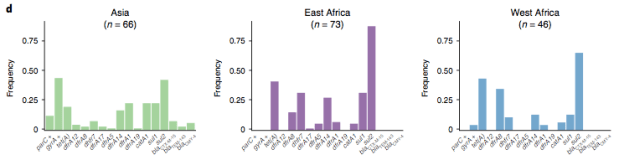
The data we have showed regional differences in AMR phenotypes, and in antibiotic usage for treatment of paediatric diarrhoea at the GEMS sites.

a) Resistance phenotypes. b) Frequency of antimicrobials prescribed to children with watery diarrhoea. c) Frequency of antimicrobials prescribed to children with dysentery.
However the prevalence of acquired resistance genes amongst E. coli isolated from each site was not associated with local frequencies of drug usage. The exception was fluoroquinolones: point mutations in gyrA and parC (which reduce MIC to ciprofloxacin) were more common at the Asian sites, where ciprofloxacin was used much more often to treat diarrheal disease than in African sites.
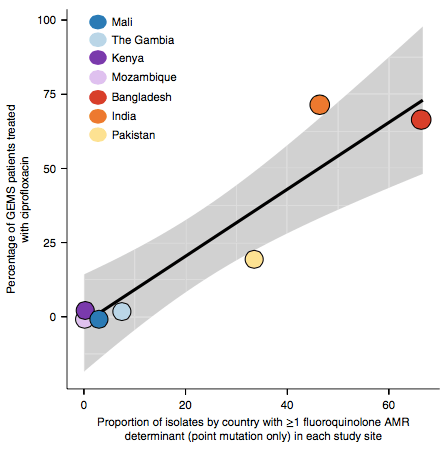
There are many possible reasons for the lack of association between local prescribing for diarrheal disease and the presence of AMR genes in local diarrheal pathogens. We expect that most antimicrobial exposure in human gut bugs like E. coli probably is not associated with attempts to treat E. coli infection at all, but with exposure to drugs given to treat other infections, drugs used in food animals which are a reservoir for E. coli, or even environmental contamination with antibiotics. Also because the horizontally acquired genes tend to travel together as a group in mobile genetic elements, exposure to one drug can co-select for resistance to many. This may be one reason that the association was more evident for ciprofloxacin use and gyrA/parC mutations, which are not in linkage with acquired AMR genes.
Finally, the data provided an opportunity to explore how well we can predict AMR phenotypes based on identifying known genetic determinants of AMR in E. coli genomes. The results were pretty good, indicating low rates of “very major errors” (where we predict a strain to be susceptible, but really it is resistant) for most drug classes. These results are comparable to those done independently in other collections of E. coli and also other bacteria, summarised here. But clearly there is room for improvement, and probably a few new mechanisms floating around out there… notably we didn’t aim to assess changes in expression of intrinsic E. coli genes, such as efflux pumps and beta-lactamases, which can contribute to drug resistance but are not so easy to find in genome data.