The first plasmid-borne colistin resistance gene was reported late last year. This was big news, but the vast majority of clinically relevant resistance to colistin and related polymixin drugs, which arises frequently in human patients being treated with these drugs, is due to de novo mutations in chromosomal genes. This has been studied quite a lot recently in Acinetobacter baumannii and Klebsiella pneumoniae, where colistin is frequently used to treat patients who are infected with carbapanem resistant strains that are also resistant to pretty much all the other antibiotics as well.
There have been quite a few studies looking for the causative mutations that underly colistin resistance in Acinetobacter and Klebsiella, by comparing the genomes of resistant and susceptible forms of the ‘same strain’ sequenced with high throughput, short read sequencing platforms like Illumina. The typical approach is to catalogue all the differences between strains and look for SNPs and other differences between them. However colistin resistance is usually associated with upregulating pmr activity via point mutations or inactivating the regulator mgrB, or by inactivating the lpx cluster in Acinetobacter. A very common way for gene inactivation to occur is by an insertion sequence (IS) hopping into the open reading frame and disrupting it. IS insertions can also cause resistance by upregulating the expression of intrinsic efflux pump or beta-lactamase genes, for example the ampC gene in A. baumannii. These insertions can be tricky to find using short read data, and are sometimes missed by regular mapping or assembly based approaches.
Luckily we now have two great tools for tracking down such mutations – ISMapper and Bandage – which were critical to tracking down polymixin resistance mutations in a recent study of A. baumannii with colleagues in Singapore. The paper is published in Antimicrobial Agents and Chemotherapy, but unfortunately is paywalled… so quick summary: basically, our clinical researcher friends in Singapore (hello Li Yang!) had 10 pairs of A. baumannii isolates, consisting of a susceptible parent isolate and a derived polymixin isolate (2 that evolved in vivo during treatment with polymixins, and 8 that evolved in vitro during polymixin exposure).
Simply screening for point mutations and deletions identified causative mutations in pmr and/or lpx clusters for 8/10 genomes, but two remained unexplained. A quick screen of the genome assemblies for IS using ISfinder identified several different IS that were present in the 10 A. baumannii genomes. However as these tend to be multi-copy in the genome, they were mostly separated into their own contigs in the assemblies. Enter ISMapper and Bandage, two open source software packages from Jane Hawkey and Ryan Wick in my lab.
First we used ISMapper to identify the locations of all IS sequences in each genome… this involves passing ISMapper each of the different IS sequences, a reference genome to compare to, and the Illumina read sets for the various strains. The isolates were all from global clones 1 or 2 (GC1, GC2), so to get the best results we used a GC1 reference genome for typing IS in the GC1 strains, and a GC2 reference for the GC2 strains. A quick tabulation of the results reveals all the locations of the various IS in each sequence. This identified differential IS insertions in lpx genes (lpxA, lpxC) in three strain pairs, including one in which no other causative mutations had been identified. There was also an IS15 insertion in the mutS gene in one isolate that had many more SNPs and deletions than the others.
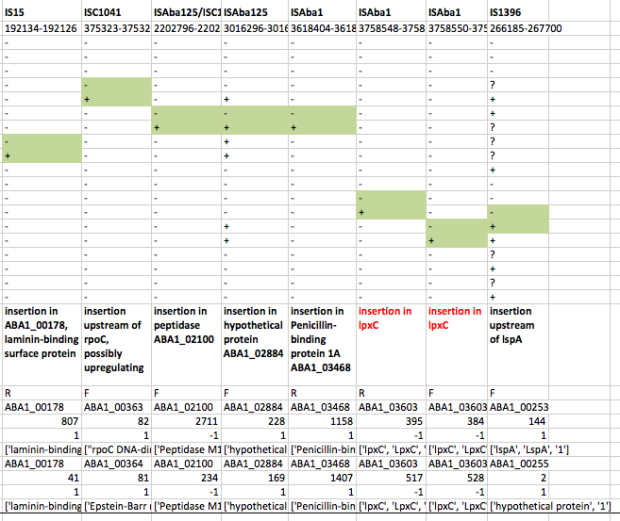
ISMapper results for GC2 strain pairs, showing IS that differed within susceptible-resistant pairs.
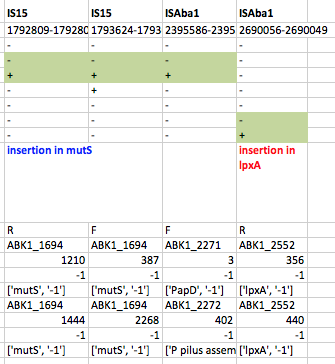
ISMapper results for GC1 strain pairs, showing IS that differed within susceptible-resistant pairs.
Cool! But what about that one last pair, where ISMapper didn’t find any differences at all between the resistant and susceptible read sets? This time we turned to Bandage, to inspect the genome assemblies and see if we could find a smoking gun. Now we had a clear hypothesis too – we were looking explicitly for interruptions in lpx genes. So we created new assemblies for these read sets using SPAdes. We loaded the graph of the susceptible isolate in Bandage first, and used the inbuilt BLAST search to locate the lpx genes within the graph – all were intact as expected, sitting happily in the middle of long contigs.

Contig containing the lpxC gene (blue) in SPAdes assembly of the susceptible isolate
Then we loaded the graph of the polymixin resistant isolate in Bandage first, and did the BLAST searches. The pmrB locus was intact, but the lpxC gene was interrupted. Very interrupted! No wonder ISMapper didn’t find this as it’s not an IS insertion at all, but rather the gene is interrupted by a large sequence, in an event that appears to involved the translocation of the entire genomic resistance island AbaR4 into the middle of the lpxC open reading frame.
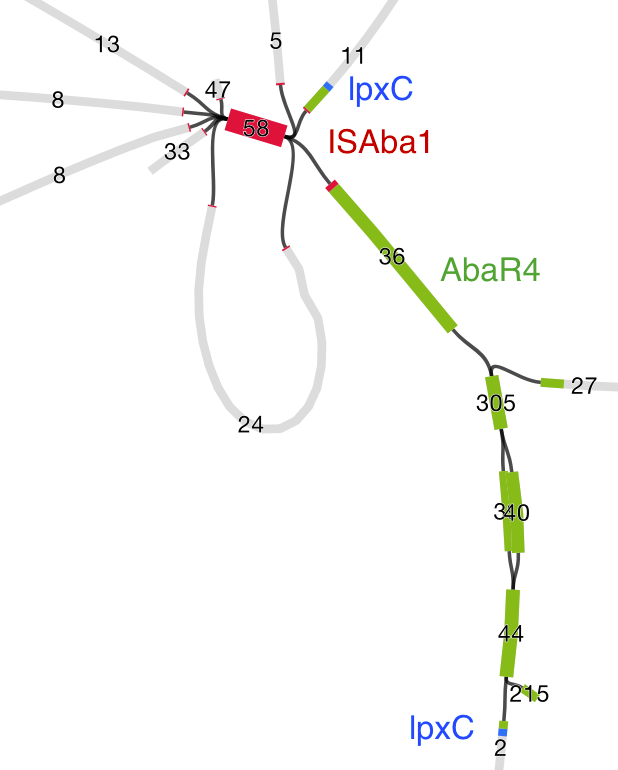
Interruption of lpxC associated with movement of the antibiotic resistance related genomic island, AbaR
The above image is created by doing BLAST searches (within Bandage) for the lpxC gene, AbaR4 gene and ISAba1 gene like this…
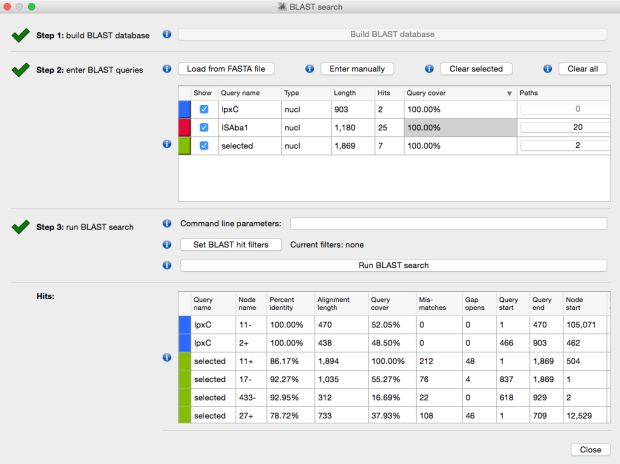
BLAST search within an assembly graph using Bandage
…and then selecting ‘BLAST hits (solid)’ under the ‘Graph display’ settings on the left hand side of the Bandage viewer.

Oh and just in case you think this is a weird one-off event that maybe you don’t need to worry about in your own genome data… check out the recent report from Scott Beaston and David Paterson in Queensland, who sequenced a nasty Klebsiella strain that was resistant to everything under the sun including carbapenems and colistin. They sequenced the genome with PacBio, and found the ISEcp1–blaOXA-181 mobile element (which confers resistance to carbapenems) inserted into the mgrB regulatory gene in the chromosome, whose inactivation is responsible for colistin resistance. Oh and they also found another mobile element, ISEcp1–blaCTX-M-15, inserted into the gene ompK35. Guess what inactivating this gene gives you? Cephalosporin resistance.