I’m often asked how we make nice annotated tree figures for our papers. The real answer is “with creativity, practice and a lot of trial and error”. But of course the answer people are looking for is what tools do we use.
There are LOADS of different tools out there for plotting trees in various ways. I’ve listed my favourites for basic tree viewing below, but these really only work for quite simple visualisations, have differing and often non-overlapping feature sets, and are hard to customise. My solution to such problems is generally to turn to Python + R, and that is exactly the case here.
As far as Python goes, I have been using the ete2 package for Python – http://etetoolkit.org/ for many years now, and thoroughly recommend it. It has a huge range of features and allows you to have a lot of control over how things look. But it requires coding in Python and a lot of referring to the manual, so a while back I wrote a wrapper script that allows you to specify a whole lot of common tree+annotation plotting features on the command line, and turns this into ete2 code to generate a PDF or png figure. Code and examples are below.
However I like to work with R and find it easier for many things, so I also wrote an R function to plot trees with data. This just uses the ape package for plotting the tree object, and other basic R functions for plotting data alongside the tree. Since then, they have released ggtree for R – http://www.r-bloggers.com/ggtree-in-bioconductor-3-1/ … this looks really great and I’m keen to try it, but since I’m comfortable with my home-grown R function and know how to deploy it in a tree-plotting emergency, I’m still using it for now.
So. In the spirit of sharing, and to save me answering the ‘how did you make that figure paper’, below is a detailed summary of the tools I use for plotting trees.
I know there are loads more out there, and this is not meant to be an exhaustive list but my personal recommendations… but if you want to share your own favourites feel free to add a comment to this post.
Annotating trees with data: Holt lab scripts
We have two work-horse scripts for plotting trees with data, one based on R (using ape) and one based on Python (using the ete2 package). Both are available in GitHub at https://github.com/katholt/plotTree and require an input tree (newick format), and take strain information or data (for heatmaps) in CSV format.
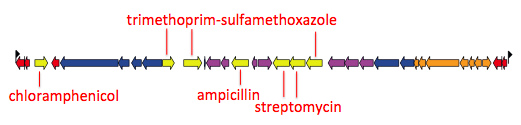
Each can do slightly different things. The biggest difference is that the R script is restricted to rectangular trees and works best for plotting associated data as text columns and heatmaps, like this example (taken from Holt et al, PNAS 2013) of a tree of Vietnamese Shigella sonnei, with tips coloured by city of isolation, and heatmap indicating the presence (black) or absence (white) of accessory genes. Example data files (newick tree + accessory gene content matrix) is available in the github repository).

A version of the R script is now included in the SRST2 repository, which can accept MLST and gene content information output by SRST2, calculate a tree from the MLST data, and plot gene content against this tree, either on an individual isolate basis or summarising gene frequencies by ST. Instructions and example data are here: https://github.com/katholt/srst2#plotting-output-in-r, including how to recreate this figure from the SRST2 paper:

The Python script is best for plotting trees in circular format with simpler, discrete categorical data, e.g. with coloured rings, or colouring branches or clades backgrounds according to tip values; it can also show branch support.
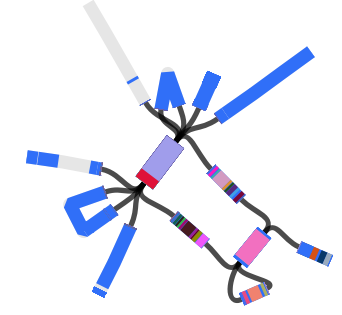
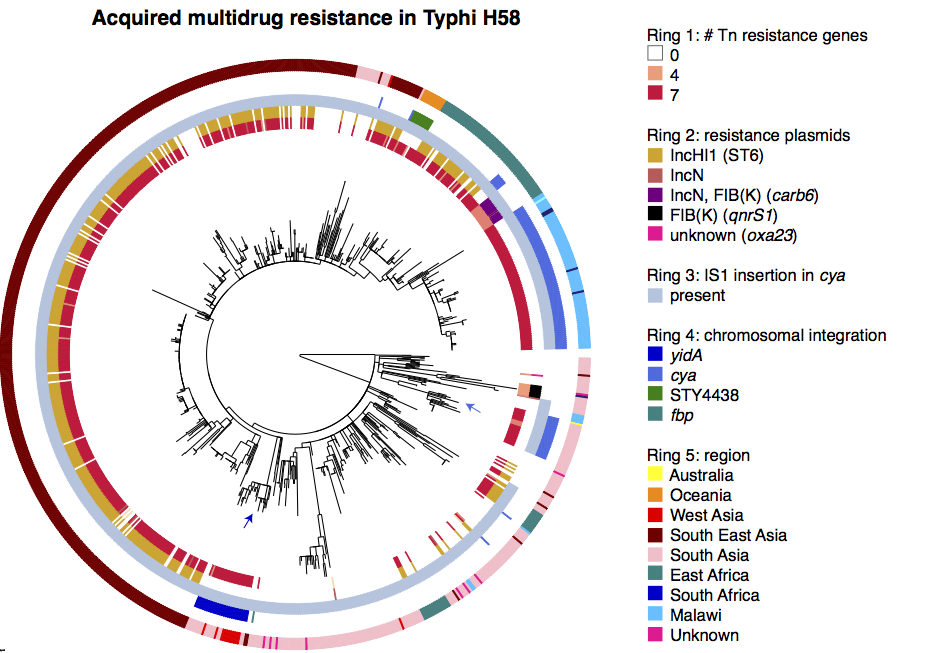
Here are some examples of the script in action, first from our Klebsiella pneumoniae paper (Holt et al, PNAS 2015)…

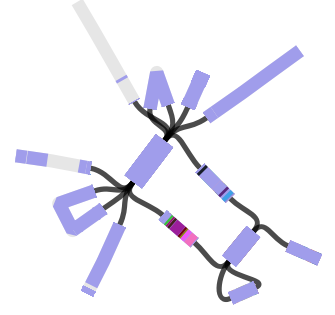
…and this example displaying MLST + serotyping data inferred from GenomeTrackr E. coli isolates, done by Danielle Ingle for her paper on serotyping of E. coli with SRST2 (manuscript in BioRxiv, serotyping instructions in SRST2 readme):

Viewing and manipulating trees (limited colouring of tree branches, tips and names only, no additional data display)
Dendroscope – http://dendroscope.org/
Dendroscope is generally my go-to for visualising, exploring and manipulating trees, although I will rarely use it to generate publication-quality figures. It has a wide range of display options (rectangular, circular, diagonal, unrooted) and allows you to very easily do things like root the tree (midpoint or on a specific branch), extract, collapse or rotate subtrees, etc. It handles input/output in a range of formats (e.g. newick, nexus) and can export images to any format (PDF, PNG, etc). Zooming of rectangular trees can be done in horizontal and vertical axes separately… this is not possible in a lot of viewers (including FigTree), but can be really important if you have big trees with lots of tips.
Tips, branches and labels can be easily coloured and manipulated, and you can save the prettied-up tree in dendroscope format for opening up later. The one thing you can’t do (I think!) is to colour clade backgrounds, which is handy sometimes and is available in FigTree.
One feature I use a lot is ‘Find’ to select tips with a particular property and then use the ‘Format’ menu to colour those tips (e.g. if I have included country in the tip names, I might search for a particular country and then colour all those tips red). The ‘Find’ dialog box has a handy ‘Find from file’ option… e.g. if I have a big tree and want to look at where a particular set of tips are distributed, I can point Dendroscope to a text file of the relevant tip names and it instantly selects those tips so that I can colour them or whatever.
FigTree – http://tree.bio.ed.ac.uk/software/figtree/
FigTree is well known as the go-to viewer for BEAST trees and it is super handy for this as it can display the estimates of height, rate, etc on each branch/node and give you a neat time axis. It is the easiest way I know of to colour clade backgrounds and to show clades collapsed into triangles with meaningful tree depths; and to colour branches by a value in the BEAST output files (e.g. rate, or posterior support). You can also load up a tab-delimited file of variables assigned to tips (column 1 = tip names, other columns = variables), and colour tips by the values of a selected variable (and also colour branches by the value of the child nodes)… although this feature seems to just not work sometimes and I can’t figure out why.
The usual views (rectangular, circular, unrooted) and manipulations (root, rotate, collapse) are available and images can be saved easily as PDF, PNG, SVG, JPG etc. However the viewer is really designed to fit the whole tree in the display window and zooming is a bit of a pain as both horizontal and vertical axes zoom together which is not always desirable (Dendroscope does better at this as explained above).
Annotating trees with data: Web-based viewers
MicroReact – http://microreact.org
MicroReact is an interactive viewer for looking at trees in the context of geography (via a map) and time (via a timeline). It can also display additional metadata via colouring tree tips and map points by the value of a single variable (you can load as many variables as you like but as they are displayed by colouring tips, you can only colour by one variable at a time).
Inputs are a tree file (newick) plus data file (CSV table), and the result is an interactive viewer with a stable URL that you can share with others (and even include in papers) in order to allow them to explore your data.
- Tree views include radial (unrooted), rectangular, circular, diagonal.
- Tree manipulations include collapsing subtrees, extracting subtrees and rotating branches.
- Tree images can be saved as PNG (with transparent background)
Interactive tree of life (iTOL) – http://itol.embl.de/
iTOL is an interactive viewer for displaying trees along with a wide range of different annotations and data types, including binary indicators, bar charts, pie charts (including on internal nodes), colour gradients, heatmaps, boxplots, animated time series, protein domain architectures and connections between nodes. It is interactive in the sense that once the data is loaded you can manipulate the tree view, and projects can be saved and shared to allow others to explore the data, however unlike MicroReact it is really most useful for creating static images.
- Trees can be viewed as radial (unrooted), rectangular or circular.
- Tree manipulations include collapsing subtrees, re-rooting, pruning and rotating branches.
- Tree images can be saved in a range of formats including PNG, SVG, PDF, etc.
ScripTree – http://lamarck.lirmm.fr/scriptree/
This is a web-based scripting system for creating static images of trees annotated in various ways, including with bar charts of data.






{kind=link}