Well our paper on “atypical” enteropathogenic E. coli is finally out, in the new journal Nature Microbiology. The hard work on this paper was done by Danielle Ingle, whom I co-supervise together with Roy Robins-Browne and will soon become the first of my students to complete a PhD. Unfortunately the paper is paywalled at the moment, so I’ll try to summarise the key elements here.
For those who aren’t familiar with the wacky world of E. coli pathotypes, the LEE genomic island is the darling of E. coli pathogenicity genetics. It encodes a type three secretion system, essentially a syringe structure that enables the bacteria to inject its own proteins (known as secreted effector proteins) directly into human cells and – ahem – mess them up (for more technical details see the impressive body of work from Melbourne University’s very own Jacqueline Pearson and Liz Hartland). E. coli that possess shiga toxin as well as the LEE cause nasty bloody diarrhoea and are defined as enterohaemorrhagic E. coli (EHEC), while those with LEE + the bfp adhesion factor cause diarrhoea and are defined as enteropathogenic E. coli (EPEC). Strains carrying the LEE but lacking both shiga toxin and bfp can also cause diarrhoea, but are also sometimes isolated from healthy individuals with no symptoms of diarrhoaea, and it is this big group of LEE + ??? strains that are defined as atypical EPEC (aEPEC), while those with bfp are dubbed typical EPEC.
No one has been able to find a common virulence factor that differentiates diarrhoeagenic aEPEC from asymptomatic strains… which is not that surprising considering that aEPEC is really just an umbrella term for a diverse group of organisms that happen to carry the LEE genomic island.
In this paper, we set out to define the population structure of aEPEC strains using genomics, and also to investigate variation in the LEE island itself. Our strain set is ~200 newly sequenced aEPEC that were isolated from children with diarrhoea, and from asymptomatic age-matched controls, as part of the Global Enteric Multicenter Study (GEMS) – a massive study into the etiology of childhood diarrhoea across seven sites in Africa and Asia, funded by the Gates Foundation. We also included lots of publicly available genomes from NCBI, which included ~60 additional aEPEC.
So what did we find? Well from a population structure point of view, we confirmed what we suspected from the beginning – that the ~200 aEPEC strains actually represent dozens of distinct lineages or clonal groups within the E. coli whole genome phylogeny. We tried making the core genome tree in a few different ways, including mapping reads to a reference genome vs using Roary to generate core gene alignments from assemblies; with and without removing recombinant regions identified using ClonalFrameML. The alternative trees all tell the same population structure story, as you can see below. An interactive version of the mapping-based, recombination-filtered tree (which appears in Figure 1 of the paper, panel a below) is available to play with in MicroReact.

The 10 clades highlighted in the tree are those containing >5 aEPEC in our collection, which represent the most common aEPEC lineages. The figures below show that these 10 aEPEC groups are present across the Asian and African GEMS sites; most also appear in non-GEMS collections from Europe as well as North and South America, indicating they are globally distributed.

Next we looked in depth at the LEE genomic island itself. ClonalFrame analysis identified lots of recombination across the locus, particularly affecting the proteins that make direct contact with host cells:
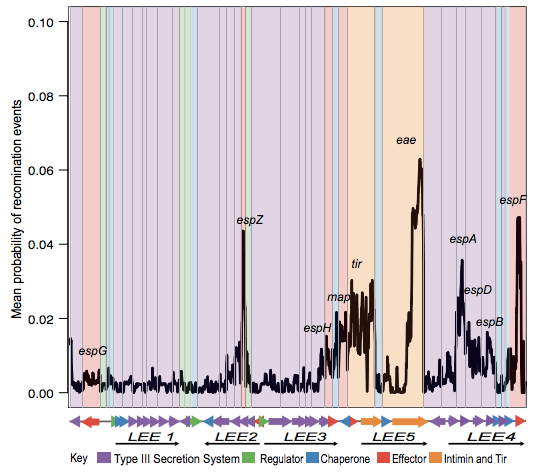
Danielle also did a lot of work to examine selection pressures on the various LEE genes, and found evidence for co-evolutionary constraints on groups of LEE genes (see the paper for details).
We used the recombination-free ClonalFrame tree, which represents the underlying clonal or vertical evolutionary relationships between LEE sequences, to define LEE lineages and subtypes. We also made a MLST-style scheme for the LEE, to make it easy to identify types from sequence data without having to compare to reference trees. The sequence files are included in the SRST2 distribution and can be downloaded from https://github.com/katholt/srst2.

There were three clearly distinct LEE lineages, separated from one other by 4-7% divergence (similar to genus-level differences between bacteria) and with different preferences for chromosomal insertion site. One of the LEE lineages (lineage 1) was entirely novel and we found it only in aEPEC isolates from the GEMS study. Functionality of the LEE was confirmed in all GEMS strains, including these novel lineage 1 examples.
Importantly, different aEPEC lineages had distinct LEE variants, often integrated at different chromosomal sites, confirming that aEPEC lineages have evolved numerous times independently via distinct LEE acquisition events. Also, some aEPEC lineages contained strains carrying different LEE lineages and/or subtypes, sometimes integrated at different sites, indicating ongoing evolution within some aEPEC clades.

Unsurprisingly, some of the “aEPEC” lineages also include examples of EHEC and/or typical EPEC isolates, i.e. subclades within the LEE-containing lineage that have subsequently acquired shiga toxin (via phage) or a bfp plasmid. We also found extensive variation in the complement of known LEE-secreted effectors (which are scattered around the genome and known as non-LEE encoded effectors or nle genes), both within and between lineages.
There’s lots more work to do to unravel the genetic basis for pathogenicity in E. coli, including identifying more of the colonisation factors, regulators and effectors that are important for causing disease in humans. But I think this analysis is a great step towards clarifying the population structure of EPEC and the LEE more generally, which opens the door for much more informative diagnostics and epidemiological studies. This work should also help immensely in guiding future pathogenicity research… most research on EPEC or EHEC pathogenicity has so far focused on a handful of strains, but our genomic analysis shows there is a much wider diversity of strain backgrounds, LEE variants and effector gene content that also needs to be considered.
