DATA: raw Illumina reads (CDC) under SRP072035
Assemblies & analyses: https://github.com/katholt/elizabethkingia
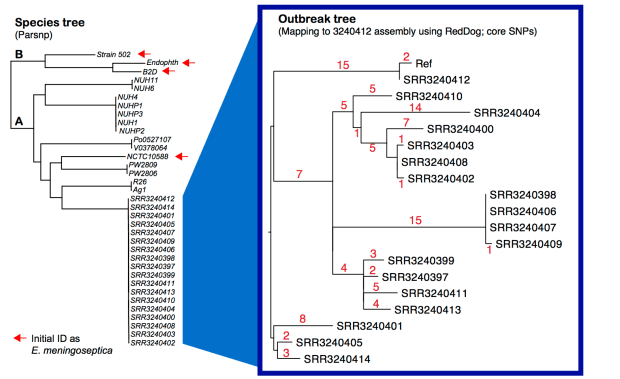
Left – core SNP tree, created from assembled genomes using Parsnp. Right – core SNP tree, created by mapping all outbreak genomes to our 9-contig assembly for SRR3240412, using our RedDog pipeline. Details below; all assemblies and mapping outputs are here.
March 19, 2016: I saw on twitter today that there was an outbreak of a weird bacteria I’ve never heard of before (Elizabethkingia anophelis) in Wisconsin, which had infected >50 people and killed almost 20.
The Wisconsin Health Services department has posted some information here (click the “For Health Professionals” tab to get info on the bacteria and antibiotic resistance).
CDC has deposited Illumina reads from 18 outbreak strains into SRA under project SRP072035 so I pulled the data and had a look. I managed to download the readsets in a few minutes (using bionode-ncbi) but it took a really long time to unpack these into fastq files using sra-toolkit.
As I have no idea about this species, I thought I’d start by looking for antibiotic resistance and plasmids in the first 6 read sets using our SRST2 software, while waiting for the rest of the reads to unpack… this ran quickly and showed me the same results for all 6 strains: GOB-10 (1 SNP from the closest allele in the ARG database) and B-2 (3 SNPs), at depths of 35-65x. For example:
SRR3240397 ARGannot.r1 GOB-1_Bla GOB-10_821 100.0 47.59 1snp 0.115 873 0.043 188 821 no;yes;GOB-10;Bla;AY647247;1-873;873 SRR3240397 ARGannot.r1 B-1_Bla B-2_1160 100.0 59.891 3snp 0.4 750 0.03 314 1160 no;no;B-2;Bla;AF189300;1-750;750
Because the matches were not identical, I pulled the consensus sequences (based on read mapping) using –report_all_consensus option in SRST2:
>314__B-1_Bla__B-2__1160 no;no;B-2;Bla;AF189300;1-750;750 ATGTTGAAAAAAATAAAAATAAGCTTGATTCTTGCTCTTGGGCTTACCAGTCTGCAGGCA TTTGGACAGGAGAATCCTGACGTTAAAATTGATAAGCTAAAAGATAATCTGTATGTATAC ACAACCTACAATACATTTAACGGGACTAAATATGCCGCTAATGCAGTATATCTGGTAACT GATAAGGGTGTTGTGGTTATAGACTGTCCGTGGGGAGAAGACAAATTTAAAAGCTTTACG GACGAGATTTATAAAAAACACGGAAAGAAAGTTATTATGAATATTGCAACACATTCTCAT GATGATCGTGCCGGAGGTCTTGAATATTTTGGTAAAATAGGTGCAAAAACTTATTCTACT AAAATGACAGATTCTATTTTAGCAAAAGAGAATAAGCCAAGAGCACAATATACTTTTGAC AATAATAAATCTTTCAAAGTAGGAAAATCCGAGTTTCAGGTTTACTATCCCGGAAAAGGA CATACAGCAGATAATGTGGTGGTATGGTTTCCAAAAGAAAAAGTATTGGTTGGAGGTTGT ATTATAAAAAGCGCTGATTCAAAAGACCTGGGGTATATTGGAGAAGCATATGTAAACGAC TGGACGCAGTCTGTACACAATATTCAACAAAAGTTTTCCGGTGCTCAGTACGTTGTTGCA GGGCATGATGATTGGAAAGATCAAAGATCAATACAACGTACACTAGACTTAATCAATGAA TATCAACAAAAACAAAAGGCTTCAAATTAA
>188__GOB-1_Bla__GOB-10__821 no;yes;GOB-10;Bla;AY647247;1-873;873 ATGAGAAATTTTGTTATACTGTTTTTCATGTTCATTTGCTTGGGCTTGAATGCTCAGGTA GTAAAAGAACCTGAAAATATGCCCAAAGAATGGAACCAGACTTATGAACCCTTCAGAATT GCAGGTAATTTATATTACGTAGGAACCTATGATTTGGCTTCTTACCTTATTGTGACAGAC AAAGGCAATATTCTCATTAATACAGGAACGGCAGAATCGCTTCCAATAATAAAAGCAAAT ATCCAAAAGCTCGGGTTTAATTATAAAGACATTAAGATCTTGCTGCTTACTCAGGCTCAC TACGACCATACAGGTGCATTACAAGATCTTAAAACAGAAACCGGTGCAAAATTCTATGCC GATAAAGAAGATGCTGATGTCCTGAGAACAGGGGGGAAGTCCGATTATGAAATGGGAAAA TATGGGGTGACATTTAAACCTGTTACTCCGGATAAAACATTGAAAGATCAGGATAAAATA ACACTGGGAAATACAATCCTGACTTTGCTTCATCATCCCGGACATACAAAAGGTTCCTGT AGTTTTATTTTTGAAACAAAAGACGAGAAGAGAAAATATAGAGTTTTGATAGCTAATATG CCCTCCGTTATTGTTGATAAGAAATTTTCTGAAGTTACCGCATATCCAAATATTCAGTCC GATTATGCATATACTTTCAAAGCAATGAAGAATCTGGATTTTGATATTTGGGTGGCCTCC CATGCAAGTCAGTTCGATCTCCATGAAAAACGTAAAGAAGGAGATCCGTACAATCCGCAA TTGTTTATGGATAAGCAAAGCTATTTCCAAAACCTTAATGATTTGGAAAAAAGCTATCTC GACAAAATAAAAAAAGATTCCCAAGATAAATAA
I had a quick look at the accessions for these genes (they are hiding in the fasta headers above) and found that they are carbapenemase genes reported from Elizabethkingia meningosepticum (previously called Chryseobacterium meningosepticum), reported in these papers: Bellais 2000, Antimicrob. Agents Chemother and Yum 2010, J Microbiol. These genes confer resistance to carbapenems like meropenem and imipenem, which probably contributes to these bacteria causing hospital-acquired infections as they will be selected for by carbapenem exposure.
SRST2 didn’t find any other acquired antibiotic resistance genes (from the ARG-Annot database) or known plasmid replicons (at least those in the PlasmidFinder database), which is consistent with the Wisconsin health services reports that these strains are susceptible to lots of readily accessible drugs including fluoroquinolones, rifampin and trimethoprim/sulfamethoxazole.
Running a quick NCBI BLAST search of the carbapenemase gene sequences shows that these new sequences, which are from outbreak strains identified definitively as Elizabethkingia anophelis by CDC, are closest to sequences annotated in NCBI as originating from Elizabethkingia meningosepticum. (The trees below are just straight out NCBI BLAST, obtained by clicking “Distance tree of results” and then downloading the newick tree files to view in FigTree.)

The outbreak strain’s GOB-10 gene had 1 synonymous SNP compared to the reference sequence, while the B-2 gene had 1 synonymous and 2 non-synonymous SNPs (affecting codons 31 & 34, which is outside the beta-lactamase domain).
I am guessing that species assignations are pretty tricky for this genus, as few labs will have access to definitive tests to discern them, so we shouldn’t read much into this. However if it is true that the outbreak strains are Elizabethkingia anophelis and the close-matching genes in NCBI did come from Elizabethkingia meningosepticum, this would suggest that there is horizontal gene transfer between these species.
Note 1: while writing this, the fastqs finished extracting and I ran SRST2 and found the same antibiotic resistance gene results on all 18. I’m now running some SPAdes assemblies which I’ll post here later, to save others the trouble…
Note 2: the assemblies (SPAdes fasta and fastg; plus Prokka annotated in GenBank format), and various analyses including trees created using Parsnp (from assemblies) and our RedDog pipeline (mapping of reads to reference genome strain NUHP1 =CP007547) are here in github: https://github.com/katholt/elizabethkingia
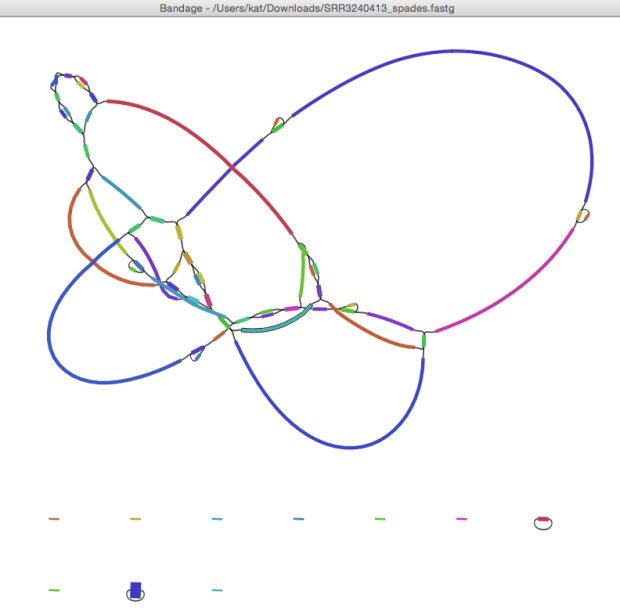
The assemblies are a bit variable, but mostly ~3.9 Mbp (the reference is 4,369,828) but the best one was for SRR3240413 – 32 contigs with 3,911,053 bp total. Viewing the SPAdes assembly graph in our Bandage program shows that 3,910,660 bp are in a single linked graph, which corresponds to the chromosome. (The other little bits do not look like plasmids, just leftover bits of sequence and probably adapters, that SPAdes spit out in teeny bits of a few hundred bp each.)
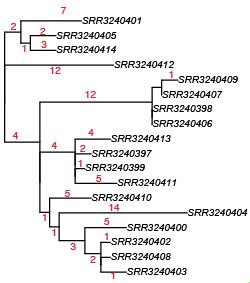
The genomes look pretty similar at first glance, but interestingly 4 of them share a deletion of ~80 genes. That’s a great little epidemiological marker for the investigations.

This was detected by our mapping pipeline RedDog, which I used to map the reads to reference genome NUHP1 CP007547 (this may not be the best reference, I just picked one randomly). The assemblies confirm it: genes BD94_0888 to BD94_0962, and the end of BD94_0963, are missing in these 4 strains (although reads do map to BD94_0948, because this is present in a second copy elsewhere in the genome).
Here’s that tree with a bit more detail (red = # core SNPs). The tree was made using FastTree, with NUHP1 reference genome as an outgroup (the outbreak strains are >40,000 SNPs away from this reference).
UPDATE 20/3
David Edwards has been playing with Hybrid StriDe + SPAdes assembly recently, and tried this with SRR3240412. The SPAdes assembly (here) was 3,913,666 bp in 41 contigs. The hybrid assembly (here) is 3,917,367 bp in 9 contigs. This is what the graph looks like… I’m showing it coloured by BLAST matches to the NUPH1 reference genome so you can see what the likely path through the graph is (rainbow, red -> purple, indicates matches from start -> end coordinates of the reference genome).

March 22: Hybrid assemblies for all 18 outbreak genomes (9-15 contigs each; ranging 3,830,044 – 3,912,928 bp) are now in GitHub (thanks to David Edwards for this).
David re-ran the RedDog mapping pipeline using this genome assembly as the reference, and got a very similar tree (files in GitHub):
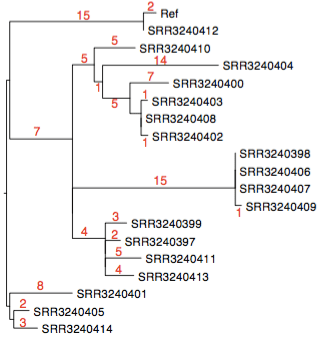
And here is a core genome SNP tree (made from genome assemblies using Harvest), which shows the outbreak strains are a novel lineage of E. anophelis, compared to currently available data (tree file in GitHub):
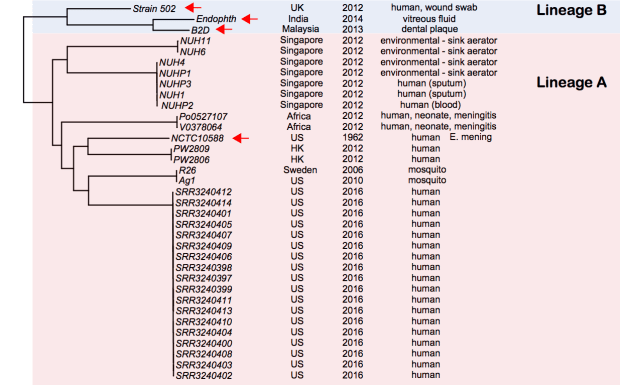
Note: Sylvain Brisse has shown the same thing using his core genome MLST scheme (see BioRxiv preprint posted March 19). I have used his nomenclature here (lineage A, lineage B). Note that Lineage B strains were originally identified as E. meningoseptica, but belong to E. anophelis. I have also included here the genome of E.meningoseptica NCTC10588, which was sequenced as part of the Sanger/PHE/PacBio type strain project, in this tree… it clusters within lineage A and is clearly E. anophelis.
UPDATE: This tweet-fest led to a collaborative project between myself, Sylvain Brisse (Institut Pasteur) and CDC, which was eventually published in Nature Communications a year after this blog post: “Evolutionary dynamics and genomic features of the Elizabethkingia anophelis 2015 to 2016 Wisconsin outbreak strain”

SRA toolkit pulls data from NCBI servers. bionode is operating on defunct access mode. you should not pull SRA data via url. it just complicates things. also, no need to ‘unpack’ to bloated fastq. SRA data structure is easily accessed more efficiently via api. Hisat reads directly from SRA 10x faster than from fastq. contact sra if you want to adapt SRST2. or check out apis here: https://github.com/ncbi/ngs
regarding metadata: the CDC and public health labs are not at liberty to reveal all details initially, but they do update when they are able.
Thanks for the comments. I will gladly try any way of downloading from SRA that is simple and less painful! Unfortunately the tools we rely on for mapping or assembly (eg bowtie2, SPAdes) need fastq as input, so I can’t see us being able to avoid unpacking SRA files for a while, it would be great though.