[Originally posted by Kat on her BacPathGenomics blog, Nov 2014]
I’m pleased to say our paper SRST2: Rapid genomic surveillance for public health and hospital microbiology labs is now out in the Genome Medicine/Genome Biology special issue Genomics of Infectious Diseases!
Having posted the code to GitHub over a year ago, and the paper to BioRxiv in June, it is great to have it finally published. I must say I love the BioRxiv experience, it is great to know the paper was viewed >1500 times while it was stuck in the peer review/publication process.
This project has been a fun journey, which started when I was lucky enough to get a fantastic summer intern, Harriet Dashnow (follow: @hdashnow), funded by VSLCI (our wonderful local supercompute cluster). Harriet’s job was to figure out how to apply our SRST code, which was designed to extract MLST information from Illumina reads (see code and BMC Genomics paper), to detect and type resistance genes and alleles. She did a fantastic job, but in doing so we realised that we needed to overhaul the scoring system to something that was faster and more robust. So, Mike Inouye designed a new scoring approach from the ground up, which Harriet and Bernie Pope from VLSCI turned into the new Python code that is SRST2. We then did a whole lot of testing on public data (mainly by Lesley Raven and Mark Schultz from the Holt & Inouye labs, with some guidance from Justin Zobel in the Computing & Info Systems department) and had our friends at the local reference lab, the Microbiological Diagnostic Unit (mainly Takehiro Tomita), test it out for themselves on a load of Listeria monocytogenes they had recently sequenced on their MiSeq.
What we came up with was some code that draws on the fundamental tools of the genomics trade (bowtie2, samtools & python) to rapidly and reliably detect genes and alleles direct from short read data. SRST2 has two modes, (i) MLST, where you can provide MLST alleles and profile definitions, and it will report sequence types; and (ii) gene detection and allele calling, suitable for screening for acquired genes such as resistance genes, virulence genes and/or plasmid genes.
Figure 1 from the paper outlines how SRST2 works:

The coolest thing about SRST2 is that it is consistent and reliable, even at low read depths, which is absolutely critical for use in a routine diagnostic/surveillance workflow. The robustness of SRST2 stems from the fact that we use a mapping approach coupled with a pretty clever approach to scoring potential matches.
The alternative is to generate de novo assemblies, and then interrogate them with BLAST or other tools… the problem with that approach is that, while de novo assemblers like Velvet and SPAdes can do a great a job of assembling bacterial genomes, there is no way to guarantee you will get a good assembly with every read set. Anyone who works with biggish bacterial data sets of 100s-1000s of genomes knows – it is very hard to standardize the assembly process and establish a protocol that works consistently and reliably every time. And if you are needing to type loads of strains as part of routine service or investigations, you need to know you will get a reliable result every time. Figure 3 in the paper compares the overall performance (call rate x accuracy) of SRST2 vs BLAST analysis of highly optimised assemblies:

As you can see, once you get up to 15-20x read depth, SRST2 will consistently give you an answer, and that answer will be accurate. Whereas using assemblies – even highly optimised ones – you will always miss out on 1-5% of the answers.
We were also asked by a reviewer to compare SRST2 with the only other publicly-available program designed for MLST typing from short reads: the MLST Typer web service at the Danish Center for Genomic Epidemiology. Because this requires uploading read sets to the other side of the world, we only did this for one of our test data sets, 44 Salmonella genomes. The results are in Table 5, and showed that the website got the correct ST for only 14/44 genomes. SRST2 got correct STs for 43/44 genomes (for the 44th strain, SRST2 returned 6/7 alleles correctly and suggested that there is actually a SNP in the 7th allele, which could actually be correct but we cannot verify; MLST Typer did not call anything for this strain).
When it comes to detecting the presence of genes, such as acquired antibiotic resistance genes, SRST2 also outperformed assembly based approaches. We got accurate detection at depths >3x, and picked up many resistance genes that did not assemble well, especially at depths below 15x (see Fig 5 of the paper). Again, we compared to ResFinder on the Center for Genomic Epidemiology website, and found SRST2 performs much better (see Table 3 in the paper). I don’t want to criticize the tools on the Center for Genomic Epidemiology website, as they are really useful for people who have limited resources to analyse genome data… but I do think people need to be aware that the successful use of those tools rely heavily on first obtaining a high quality assembly, and you can very easily miss important antibiotic resistance genes.
So what are the main uses for SRST2?
The key jobs SRST2 is good for are:
- detecting the presence of genes
- identifying specific alleles for those genes from a known set of sequencescalculating sequence types (STs) specified by MLST-style schemas.
So it is great for detecting acquired genes, such as antibiotic resistance genes, virulence genes or plasmid genes; or extracting MLST information. To this end, the SRST2 download includes:
- a script to help you pull down the latest MLST data from pubmlst.org for >100 MLST schemes
- instructions and code for typing virulence genes from the Virulence Factor Database (VFDB)
- SRST2-compatible versions of popular resistance gene databases (ARG-Annot, ResFinder) and the PlasmidFinder database of plasmid replicons
For example, in the paper we show the use of SRST2 for investigating changing patterns of vancomycin resistance amongst Enterococcus faecium isolated at a local hospital (data from this 2013 MBio paper). We analysed the Illumina readsets using SRST2 to detect resistance genes and perform MLST. SRST2 returned a table of strains with columns to indicate sequence types, alleles for each locus in the MLST scheme, and the presence of acquired resistance genes (shown here viewed in Excel):
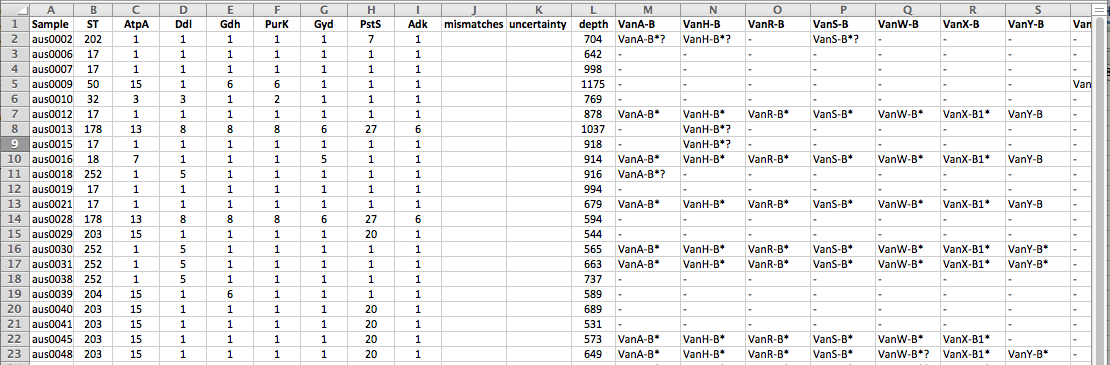
You will notice that the depth values are VERY high for this data set, as the sequencing was done at relatively low-plex on a HiSeq. This means that it takes a long time to map (and VERY long time to assemble) these read sets… however note that you can obtain the same results much faster by telling SRST2 to use just the first 1 or 2 million reads per strain, which is more than enough to get accurate results (–stop_after 1000000).
We used R to summarize and plot the results, which are shown in Figure 7 in the paper (R code is in the /scripts directory in the SRST2 download):

Panel (a) shows that vanB vancomycin resistance became more frequent over the 12 years investigated, while panel (b) shows that the STs also changed during this time period… so is the increase in resistance linked to the introduction of new van-resistant ST252 and ST203 strains?
Panel (c) puts the ST & resistance results together… the tree on the left is a dendrogram depicting the similarity between sequence types based on number of shared alleles; MLST profiles (STs and their component alleles) are shown next to this. On the right, the frequency of each ST is shown as a bar graph, which highlights that there are 3 main types, ST17, ST252 and ST203, which all share 5-6 alleles. The heatmap in the middle shows the frequency of the various resistance genes within each ST – it is clear that the vanB locus is present in all 3 STS, but only at ~50% in each… so the increase in vancomycin resistance is not explained by the introduction of resistant ST252 and ST203 clones; rather, vancomycin resistance comes and goes, via movement of the vanB operon, in all 3 common STs.
The whole genome phylogenetic analysis in the 2013 MBio paper shows this in much finer detail of course; but the basic conclusions could be drawn from a very rapid analysis with SRST2.
The same is true for hospital outbreak investigations (see examples in the paper) – in a few minutes SRST2 can show whether a potential transmission case has the same ST and acquired genes profile as confirmed outbreak cases; if the ST matches, genome-wide SNP trees can be used to investigate further.
My next post will have some pro tips for using SRST2.
